Le rôle des mitochondries
(Le Petit A.M.Mi N° 2 Décembre 1999)
Dr Pierre RUSTIN
La présentation qui suit sera la plus informelle possible : son but sera d’essayer de vous faire comprendre » en gros » à quoi sert une mitochondrie dans une cellule, mais aussi ce qu’est une cellule.
Les éléments les plus » à la base » possible seront exposés, afin que vous puissiez suivre toutes les données qui vont vous être apportées au cours de cette après-midi consacrées aux maladies mitochondriales par l’ensemble des intervenants. N’hésitez pas à poser des questions ; le seul objectif de cette réunion, c’est que vous appreniez des choses qui sont nécessaires pour comprendre les maladies mitochondriales.
Pour débuter, il faut indiquer que les mitochondries se trouvent dans une cellule.
Le corps humain est constitué de millions de cellules. Toutes les cellules sont construites sur le même schéma, bien qu’elles ne soient pas toutes identiques. Ce schéma d’une cellule va vous être présenté, de façon à ce que vous puissiez placer les mitochondries à l’endroit où elles sont.
Une cellule peut mesurer par exemple 50 microns (le micron correspond à 1 millimètre divisé par mille, pour vous donner une idée de la taille de ces cellules). Certaines autres cellules peuvent mesurer des centimètres, voire des mètres. Toutes ces cellules sont construites de la même façon.
Une cellule est d’abord séparée de son environnement par une membrane, appelée membrane plasmique.
Si l’on schématise cette cellule, par exemple, une cellule de la peau (c’est-à-dire un fibroblaste, dont on se sert pour faire un diagnostic de maladie mitochondriale), on trouve d’abord un compartiment, qui est séparé de l’extérieur par la membrane plasmique. » Séparé » n’est pas un terme adéquat, car c’est au niveau de cette membrane que se font les échanges entre la cellule et l’extérieur.
A l’intérieur de la cellule, on trouve un gel riche en eau, appelé cytoplasme. Dans ce gel, il se déroule des millions de réactions chimiques d’une complexité extraordinaire, et qui ont pour fonction d’assurer la survie et la reproduction de la cellule. Ces réactions forment ce qu’on appelle le métabolisme. Les maladies mitochondriales sont des maladies du métabolisme, liées à des dysfonctions de celui-ci.
Dans chaque cellule, on trouve une information héréditaire (c’est-à-dire héritée des parents), sous la forme de molécules appelées ADN, qui sont contenues dans le noyau. Ce noyau est entouré d’une membrane perméable (le noyau communique avec le reste de la cellule) et renferme l’information dite « génétique » (les gènes) sous la forme de filaments d’ADN organisés de façon à contenir cette information ; l’unité d’information est appelée » gène « , et un ensemble de filaments d’ADN constitue un chromosome.
Le noyau contient donc des chromosomes, qui vont en général par paire, et sur ces chromosomes, on trouve les gènes.
La copie d’un gène peut sortir du noyau (au travers de sa membrane perméable) sous la forme d’un ARN ; cet ARN, une fois transporté à l’extérieur du noyau, sert à construire, par traduction, le plus souvent une protéine ; cette protéine est en quelque sorte l’ouvrier de la cellule : elle participe aux réactions chimiques intra-cellulaires évoquées précédemment.
Quand existe une maladie génétique, une des causes possibles peut être une anomalie de fonctionnement du gène, qui est recopiée au niveau de l’ARN, et la protéine finalement produite fonctionne mal ; une fonction de la cellule est donc mal assurée (par exemple, la protéine qui transforme une substance A en B ne sera plus capable de fabriquer B). Cela représente un premier type de maladies du métabolisme.
Dans la cellule, il existe d’autres constituants, en particulier
–des filaments, qui structurent la cellule, lui donnent sa forme et organisent le mouvement de tout ce qui se trouve dans la cellule ;
–de petits éléments (organites ou organelles), tels que :
–les mitochondries, souvent filamenteuses et ayant tendance à former un réseau ; elles fabriquent l’énergie de la cellule ;
–les peroxysomes, petits sacs qui permettent à la cellule d’effectuer des réactions extrêmement dangereuses, en autorisant la manipulation de substances très toxiques (les peroxydes) ;
–le réticulum endoplasmique, formé de sacs aplatis au niveau desquels sont fabriqués la plupart des constituants de la cellule, ainsi que des éléments qui vont sortir de la cellule ;
–les lysosomes, véritables » poubelles » de la cellule, sièges de destruction des éléments indésirables de la cellule.
Ces éléments sont en général présents dans toutes les cellules, mais en proportions variables.
Les mitochondries :
Leur nombre au sein d’une cellule est extrêmement variable en fonction des besoins de celle-ci en énergie, allant de quelques centaines à près d’un million. Par exemple, dans un muscle fréquemment sollicité, qui travaille donc souvent, leur nombre a tendance à augmenter (car la demande en énergie est élevée).?????…/…
Le principal problème des maladies mitochondriales est donc un problème d’énergie.
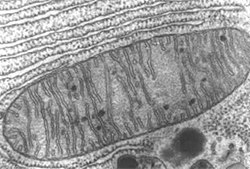
La mitochondrie peut-être représentée sous la forme d’un sac qui a deux membranes :
– une membrane externe, qui est perméable,
– une membrane interne, très plissée, qui est le siège de la synthèse (= fabrication) de l’ATP.
A l’intérieur de la mitochondrie, il y a un gel particulièrement dense ; lorsque la mitochondrie fonctionne, elle prélève dans le cytoplasme des cellules des composés qu’elle peut consommer, tels le pyruvate, et va les concentrer dans ce gel. Le pyruvate y est dégradé par un cycle de réactions chimiques (le cycle de Krebs), qui va le fragmenter ( » casser « ) en constituants plus petits. A chaque fragmentation, de l’énergie est libérée, et cette énergie est employée pour fabriquer de l’ATP.
Incidemment, si la mitochondrie ne peut pas utiliser le pyruvate (par mauvais fonctionnement), celui-ci ne va pas pouvoir entrer dans la mitochondrie et va se transformer en lactate, d’où les acidoses lactiques souvent évoquées lors de maladies mitochondriales. Ceci permet de réaliser un test diagnostic : des concentrations anormales de lactate dans les liquides de l’organisme peuvent être une indication d’un dysfonctionnement des mitochondries (cet indicateur n’est pas toujours présent, car il dépend de la quantité de cellules et d’organes atteints).
La production de l’ATP :
Elle se fait au niveau de la membrane interne de la mitochondrie, lieu où se situe la chaîne respiratoire, système cellulaire de récupération de l’énergie.
La chaîne respiratoire est formée de 5 grands complexes enzymatiques ; ils sont identifiés par un numéro, de 1 à 5 (CI à CV).
Lorsque les molécules de pyruvate se fragmentent, sont cassées dans la mitochondrie, l’énergie en résultant est transmise au premier complexe de la chaîne respiratoire sous la forme d’un électron : l’énergie contenue dans les liaisons chimiques est donc transformée en énergie électrique (électron) au niveau de la membrane interne. Ces électrons qui transitent dans la membrane interne vont aboutir à l’oxygène, s’y accumuler et produire de l’eau. C’est le transit de ces électrons qui va représenter l’énergie utilisée pour fabriquer de l’ATP au niveau du 5ème complexe de la chaîne respiratoire.?
Question : » On parle de maladie génétique pour les maladies mitochondriales, pourtant vous nous dites que les gènes sont dans le noyau … Quel rapport avec la mitochondrie ? «
R : La plus grande partie des ARN est synthétisée à partir des gènes du noyau ; les protéines sont, elles, synthétisées à partir des ARN. Ces protéines vont être distribuées dans tous les compartiments de la cellule, y compris dans les mitochondries. De ce fait, les mitochondries peuvent être déficientes, si un des gènes présents dans le noyau fonctionne mal. Parce que ce gène sera » muté « , l’ARN sera lui aussi » muté « , la protéine en résultant ne sera pas fonctionnelle, et elle sera envoyée, avec son problème fonctionnel, dans la mitochondrie, et la mitochondrie fonctionnera mal. Ces maladies mitochondriales sont dites » nucléaires « , car le gène responsable est présent dans le noyau de la cellule.
Les protéines mitochondriales ont aussi une seconde origine : elle se situe dans les mitochondries, qui contiennent aussi un petit nombre de gènes (A. Rötig abordera ce point).
Question : » Pourrait-il y avoir des maladies mitochondriales non génétiques ? »
R : Elles peuvent se rencontrer, car il existe des toxiques qui peuvent léser les mitochondries. Mais celles que nous étudions sont en général génétiques.
L’autre » catégorie » de maladies génétiques évoquées (celles qui ne sont pas » nucléaires « ) ont pour origine une anomalie d’un gène de l’ADN mitochondrial, car les mitochondries possèdent leur propre matériel génétique.
Il y a donc deux classes de maladies mitochondriales :
– celles d’origine nucléaire,
– celles d’origine directement mitochondriale.
Question : » Qu’appelez-vous mutation d’un gène ? »
R : Un gène est une unité d’information, qui peut être considéré comme un mot très long constitué de lettres;
cet enchaînement de lettres utilise un alphabet qui aurait 4 lettres en tout, et il suit un ordre fixe, et c’est cet ordre qui contient l’information. Pour que le gène soit fonctionnel, il faut que l’ordre soit strictement respecté.
Une maladie génétique, c’est une maladie dans laquelle, pour une raison quelconque, cet ordre est perdu. Ce peut être :
-une lettre (ou plusieurs) échangée(s) avec une (des) autre(s),
-une lettre (ou plusieurs) qui manque(nt),
-une lettre (ou plusieurs) qui se rajoute(nt).
Tout ce qui va toucher l’ordre des lettres va entraîner un dysfonctionnement. C’est ce qu’on appelle une mutation.
Question : » Quelle est la durée de vie d’une mitochondrie ? Est-ce qu’elles se renouvellent ? «
R : A chaque division cellulaire, le stock des mitochondries de la cellule-mère est divisé en deux. Dans les tissus où les cellules ne se divisent pas ou peu, les éléments de la cellule se renouvellent régulièrement. Il est difficile d’indiquer un rythme précis de renouvellement des mitochondries : certains de ses éléments vont se renouveler plus vite que d’autres, et il s’agit en fait d’un renouvellement permanent.
Les mitochondries (suite) :
Elles sont toutes organisées de la même façon, et proviennent toutes, il y a très longtemps, d’une bactérie qui a permis à la cellule d’utiliser l’oxygène.
Le rôle essentiel de la mitochondrie est en effet d’utiliser un combustible, de le » brûler » avec de l’oxygène, de récupérer l’énergie dégagée par cette combustion et de fabriquer de l’ATP à partir de celle-ci.
Lorsqu’il existe un déficit de la chaîne respiratoire, un des complexes de celle-ci peut-être touché : le flux des électrons dans la membrane est ralenti, et donc la synthèse d’ATP est ralentie.
Ce problème est d’autant plus compliqué que, par exemple, le complexe I est lui-même formé de 40 composés différents intervenant dans les réactions pour faire progresser les électrons dans la membrane. Chacun des complexes de la chaîne respiratoire est ainsi formé de plusieurs sous-unités, ce qui donne une idée de la complexité du système. Il y a ainsi au moins 100 protéines différentes impliquées dans la chaîne respiratoire, ce qui fait donc intervenir au moins 100 gènes différents (essentiellement des gènes du noyau de la cellule, mais pas exclusivement : 13 sont des gènes de l’ADN mitochondrial).
Question : » Vous avez dit tout à l’heure que si quelque chose ne marche pas, le pyruvate est transformé en lactate. D’où vient le pyruvate ? »
R : Dans une cellule normale, à partir des sucres, le glucose est fragmenté en divers éléments et aboutit au pyruvate. Lorsque la mitochondrie est fonctionnelle, la plus grande partie du pyruvate entre dans les mitochondries, où il est dégradé pour produire de l’ATP. Si le système qui produit l’ATP n’est pas fonctionnel (déficit de la chaîne respiratoire), le pyruvate ne peut pas être consommé par les mitochondries, et il est transformé en lactate à l’extérieur de la mitochondrie, avant de sortir de la cellule pour passer dans les liquides de l’organisme où il peut être dosé.
Question : » Quand la mitochondrie dysfonctionne, c’est l’ensemble des mitochondries du corps humain qui dysfonctionnent, ou ce sont seulement quelques-unes ? »
R : Cela dépend du type de dysfonctionnement, et de son origine génétique. S’il s’agit d’une maladie mitochondriale nucléaire, toutes les mitochondries, de façon théorique, sont touchées. A l’inverse, si ce sont des maladies mitochondriales par atteinte de l’ADN mitochondrial, certaines mitochondries sont touchées, d’autres pas.
Le problème est en fait plus complexe que cela. Dans certaines maladies mitochondriales nucléaires, des phénomènes, que nous ne comprenons pas, font que certains organes sont touchés, et d’autres pas.
Question : » Pourquoi ne peut-on pas absorber de l’ATP pour compenser le déficit des mitochondries dans les maladies mitochondriales ? »
R : On ne peut pas, car il est détruit instantanément. Une des approches de ces maladies pourrait être d’employer des constituants qui soient capables de générer plus d’ATP dans la cellule.
Autres rôles de la mitochondrie :
A côté de cette fonction liée à la production d’ATP, la mitochondrie a d’autres rôles qui font, en cas de dysfonctionnement, apparaître d’autres problèmes. La mitochondrie intervient ainsi dans le contrôle de la mort cellulaire. Il est donc possible que, dans certaines maladies mitochondriales, il y ait un dérèglement de ces fonctions de contrôle, à l’origine de nécroses tissulaires, ou au contraire, de proliférations cellulaires anormales.
Les fonctions de la mitochondrie sont donc très variées, et ne sont pas centrées que sur la production d’ATP.
Quelques approches thérapeutiques des atteintes de la chaîne respiratoire :
Il est classique de comparer la mitochondrie a une usine électrique ; il serait mieux de dire une » centrale nucléaire « , car dans la mitochondrie, il y a des éléments particulièrement dangereux.
Ainsi, dans la membrane interne de la mitochondrie, il y a des électrons qui transitent comme dans un fil électrique ; ce fil électrique n’a pas de » gaine « , ce qui revient à dire que s’il y a » trop de courant « , des électrons vont s’en échapper. Les électrons isolés de ce type sont très agressifs, et ils détruisent tous les composés qu’ils rencontrent : c’est ce qu’on appelle les radicaux libres (en fait, une molécule avec un électron qui se » promène seul « ). La mitochondrie, lorsqu’elle fonctionne mal, en produit beaucoup. Une approche thérapeutique peut se faire ici, dans le but de contrôler la quantité d’électrons qui s’échappent de la chaîne respiratoire.
Une seconde approche est envisageable. Dans le transfert d’électrons au sein de la membrane interne de la mitochondrie, certaines étapes de la chaîne respiratoire peuvent être interrompues par absence, manque d’un composé. Exemple avec l’ubiquinone : en réintroduisant ce composé déficitaire dans l’organisme, par diffusion jusqu’aux mitochondries, on arrive à restaurer le transfert déficient des électrons. On recherche actuellement en laboratoire d’autres composés qui, comme l’ubiquinone, permettraient de contourner, de » shunter » les étapes qui ne fonctionnent pas ou mal.?